Nuevo método de construir árboles filogenéticos
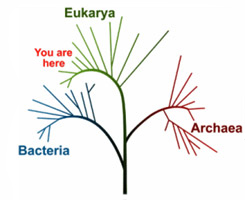
Un sistema algorítmico puede, a partir de la información genética, medir distancias evolutivas para ciertas proteínas y reconstruir árboles filogenéticos.
|
Hasta hace poco reconstruir los pasos evolutivos que llevaron de una especia a otra o la filogenia de las especies era muy laborioso y difícil. Comparar rasgos anatómicos o buscar fósiles en las entrañas de la tierra de un lugar remoto eran algunas de las tareas que se necesitaban realizar. Ahora, además de esos métodos imprescindibles, contamos con poderosas computadoras y secuenciadoras genéticas. Podemos leer en el «libro genético» que cada ser vivo contiene para saber cómo evolucionaron y se relacionan entre sí todas las especies. Allí podemos bucear millones de años atrás en el tiempo para, en el fondo, saber un poco más de nuestros origen, y por tanto, de nosotros mismos.
Se han desarrollado varios de estos métodos, y probablemente se desarrollen otros en el futuro. El que ahora presenta un grupo de Penn State University es un método nuevo que ayudará a entender el origen, evolución y diversidad de la vida en la Tierra. Se podrá estudiar la historia evolutiva de las proteínas atrás en el tiempo y quizás ayude a aclarar qué formas de vida surgieron primero sobre este planeta. El nuevo método se publica en Proceedings of the National Academy of Sciences.
El método se centra en un grupo de proteínas denominadas retroelementos, que componen, por ejemplo, aproximadamente el 50% en peso el genoma humano. Juegan un papel crucial en muchos procesos incluido el de la infección por el virus del SIDA.
Los retroelementos son una antigua y diversa clase de proteínas, por tanto pueden proporcionan un riguroso punto de referencia sobre el que comprobar esta aproximación.
Este grupo de investigadores ha desarrollado los algoritmos necesarios para hacer que el método funcione y los publica libre y gratuitamente en forma de software open-source para que otros investigadores lo puedan descargar de la red y utilizarlo.

|
|
|
Los científicos pueden estudiar las historias evolutivas de las distintas especies mediante la comparación de las secuencias genéticas. Los organismos más cercanos evolutivamente a otros que comparten un ancestro común tienen secuencias similares.
En el artículo (descargable de la web PNAS a partir del día 6) se describe el estudio de 11 grupos de retroelementos y se traza sus historias evolutivas. El método usa los algoritmos implementados sobre programas de ordenador para generar perfiles evolutivos, también llamados perfiles filogenéticos, que son comparados entre sí. De este modo encuentran las regiones de los perfiles que encajan entre ellas para crear así un diagrama en forma de árbol filogenético. El árbol proporciona la distancia evolutiva, es decir, las relaciones filogenéticas entre retroelementos.
En los métodos anteriores también se podía conseguir esto mismo, pero era necesario una tarea manual por parte del investigador en algún momento. Aunque la mente humana es muy poderosa a la hora de reconocer patrones, estos procesos eran lentos laboriosos.
Asimismo el nuevo método puede utilizase en conjunción con estos métodos previos para obtener unas historias evolutivas más claras y fiables.
El nuevo método también sirve para hallar las características funcionales y estructurales de las proteínas a la vez que se miden distancias evolutivas.
Hay unos 30.000 perfiles en los repositorios científicos a disposición de todos los científicos que pueden ser usados para generar perfiles filogenéticos. Los investigadores del grupo esperan que el método sea más preciso según se añadan nuevos perfiles al banco de datos, ya que es lo que ha estado pasando hasta el momento. Ahora ya producen árboles filogenéticos más precisos de los que aparecen en su artículo.
Su meta es trazar estas historias evolutivas hasta el origen de la vida sobre la Tierra.
Fuentes y referencias:
Noticia en Penn State.
Nota de prensa en Penn State.
Resumen del artículo (abierto) en PNAS.

5 Comentarios
RSS feed for comments on this post.
Lo sentimos, esta noticia está ya cerrada a comentarios.






lunes 8 septiembre, 2008 @ 3:27 pm
«El método se centra en un grupo de proteínas denominadas retroelementos».
¿Los retroelementos no son una familia de genes, que emplean mediadores de ARN para reproducirse nuevamente como ADN?
«En los métodos anteriores también se podía conseguir esto mismo, pero era necesario una tarea manual por parte del investigador en algún momento. Aunque la mente humana es muy poderosa a la hora de reconocer patrones, estos procesos eran lentos laboriosos.»
También se ha utilizado para estudiar relaciones evolutivas, diferentes secuencias de arn ribosómico que al cumplir una función muy importante se han conservado a lo largo de la evolución, por lo tanto solo hay que comparar las secuencias de estos ARNr contenidos en las bibliotecas para trazar arboles evolutivos muy precisos, por lo tanto es falso que haya que hacer un trabajo manual, en todo caso el mismo que habría que hacer ahora para construir las bibliotecas de secuencias de nucleótidos de los retroelementos, aunque todo método nuevo para trazar arboles evolutivos es muy interesante en la medida que sirven para comparar con los ya existentes y ver si coinciden.
Y además no creo que nadie utilice un proceso manual para comparar secuencias de adn, los ordenadores están para algo y los algoritmos para esto no creo que tengan gran dificultad.
Saludos.
lunes 8 septiembre, 2008 @ 7:51 pm
En internet se pueden leer definiciones sobre qué son los retroelementos. Esta es una de ellas:
Desconocemos si se refiere a los que se mencionan en este trabajo. Habría que leer el artículo original en detalle.
Por otro lado la capacidad de apreciar patrones del cerebro humano no ha sido superado ni igualado por ningún programa. Por eso esta misma web tiene una serie de números que usted debe de introducir para comentar y que un programa robot no sabe reconocer.
El ADN
ribosómicomitocondrial se ha empleado y se emplea para este tipo de asuntos, pero al ser más pequeño no aporta tanta información como el ADN nuclear. Los resultados obtenidos con él son bastante discutibles y entre los expertos suele haber polémica. Recuérdese el caso de la Eva negra, por ejemplo. ¿Para qué conformarse con una parte de la información pudiendo tener mucha más, concretamente la parte que determina el fenotipo?Por cierto, se ha añadido un enlace al artículo original, que además está «en abierto».
martes 9 septiembre, 2008 @ 12:14 am
«Por otro lado la capacidad de apreciar patrones del cerebro humano no ha sido superado ni igualado por ningún programa.»
La idea para saber si dos organismos están emparentados es ver si las secuencias se conservan, es decir si son las mismas en dor organismos diferetes que queramos comparar, y esto lo hace muy bién un ordenador.
«El ADN ribosómico se ha empleado y se emplea para este tipo de asuntos, pero al ser más pequeño no aporta tanta información como el ADN nuclear.»
Que yo sepa en los ribosomas no hay ADN, sino ARNr y proteinas.
Saludos.
martes 9 septiembre, 2008 @ 8:56 am
Tiene razón, un confusión con el ADN mitocondrial imperdonable. Ya ha sido corregido.
En cuanto a la participación humana en otros métodos sólo se hace en determinadas ocasiones, el trabajo en bruto lo hace un ordenador. En la nota de prensa se dice:
«To obtain more detailed information about possible relationships among the sequences, a human expert who can manually search for such relationships is needed.»
martes 9 septiembre, 2008 @ 10:22 am
Parece lógico que al final sea una persona la que decida si existe relación filogenética entre dos organismos que está estudiando y estudie con con más detalle los organismos que el ordenador clasifique como candidatos a estar emparentados.
Saludos.