Consiguen deducir la estructura de las proteínas
Consiguen deducir computacionalmente la estructura terciaria de las proteínas a partir de la secuencia de aminoácidos.

|
|
|
Una proteína no es más que una secuencia lineal de aminoácidos que viene determinada por la secuencia de bases de ADN del gen que determina dicha proteína. Pero esa secuencia de aminoácidos se auto-organiza y se produce lo que se llama el plegamiento de la proteína. La forma final que adopta está determinada específicamente por esa secuencia de aminoácidos, pero también depende del medio en el que se encuentre.
La función de una proteína viene dada por esa estructura tridimensional. Si se cambian algunos aminoácidos se cambia la estructura terciaria de la proteína y, por tanto, su función. Hay pequeños cambios en esa secuencia que no producen cambios en la función de la proteína y que permite cierto margen de error a la evolución.
Para desarrollar un nuevo fármaco muchas veces se necesita saber la estructura tridimensional final de una proteína, pero para saber esa forma se requieren procesos de cristalografía por rayos X o resonancia magnética nuclear que son largos, laboriosos y caros. El sueño sería meter la secuencia de aminoácidos (o la correspondiente secuencia de bases de ADN, pues esta relación es biunívoca) en un computador y que éste calcule esa forma. Esta meta se ha perseguido durante décadas sin mucho éxito hasta el momento. Sólo se podían predecir algunos casos sencillos. Esto se debe a que el número de las posibles configuraciones en las que esos aminoácidos pueden colocarse en el espacio es enorme. Es básicamente un desafío combinatorio.
Ahora, cincuenta años después de que se descubriera que la estructura tridimensional de las proteínas determinaba su función, se ha logrado dar un gran paso adelante en la predicción computacional de esa estructura de las proteínas. El logro lo ha realizado un equipo internacional formado por varios grupos de investigadores y es publicado en PLoS ONE.
La determinación computacional de esta estructura entraña una enorme complejidad y sin algún tipo de atajo se torna imposible que un moderno computador la pueda determinar en un plazo de tiempo razonable. Pero ya hay mucha información genética disponible, y muchas informaciones, técnicas y aproximaciones al problema, así que la clave está en la colaboración entre distintos especialistas en distintos campos: Genética, Matemáticas, Biología Evolutiva, etc.
Estos investigadores partieron de una premisa: que la evolución podría proporcionar una guía a la hora de determinar el plegamiento de la proteína. En su aproximación emplean tres elementos clave: la información evolutiva acumulada durante millones de años, los datos sobre secuenciación de genes conseguidos durante estos años y un método de Física Estadística recientemente desarrollado.
|
La información genética acumulada en la forma de miles de proteínas permitió agrupar a las mismas en familias que tienen formas similares. Un algoritmo les permitió inferir qué partes de la proteína interaccionan para determinar la forma final. Entonces usaron un principio de la Física Estadística, denominado de máxima entropía, que extrae información acerca de la interacciones microscópicas a partir de medidas sobre la propiedades del sistema.
Este método resultó ser sorprendentemente efectivo a la hora de extraer información esencial a partir del registro evolutivo.
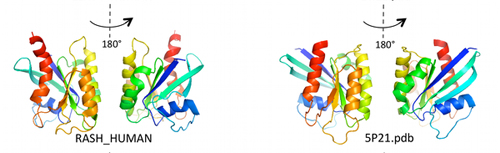
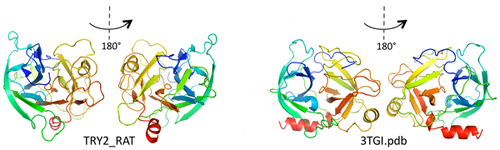
A partir de esta información sobre las interacciones internas de la proteína, este grupo de investigadores usó un software de simulación molecular para generar la forma de la proteína a escala atómica. Es la primera vez que a partir de secuencias se computa con tan notable exactitud una proteína, algo que han conseguido en 15 casos distintos sin demasiadas limitaciones en tamaño sobre proteínas cuya estructura ya se conocía de antemano experimentalmente. Incluso han podido resolver la estructura de proteínas de la familia Ras de unos 160 aminoácidos. Pero no hay razón para que el método funcione igualmente para proteínas mayores.
|
Al parecer, a nadie se le ocurrió poner juntos todos estos métodos y datos ya existentes para determinar las estructura 3D de las proteínas, pero funciona muy bien. El error cometido en la localización espacial de los elementos estructurales es menor de 3,5 armstrongs respecto a lo que se sabe por cristalografía de rayos X.
El próximo paso de estos investigadores es predecir la estructura de proteínas para las que no hay datos de cristalografía de rayos X, pero que ya están investigándose, antes de adentrase en un terreno completamente desconocido.
Quizás dicho así, y dejando aparte el autobombo de la institución de turno, el resultado no parezca importante. Pero si es verdad que si el método puede escalarse y aplicarse a otros casos se trataría de un logro revolucionario en el campo.
Copyleft: atribuir con enlace a http://neofronteras.com/?p=3687
Fuentes y referencias:
Nota de prensa.
Artículo original.

2 Comentarios
RSS feed for comments on this post.
Lo sentimos, esta noticia está ya cerrada a comentarios.






viernes 16 diciembre, 2011 @ 9:49 am
Quizás no lo parezca, pero las medicinas del futuro dependerán de este estudio o de estudios como éste.
lunes 19 diciembre, 2011 @ 10:15 am
Por eso, en todos los campos la investigación básica es imprescindible.